Overview
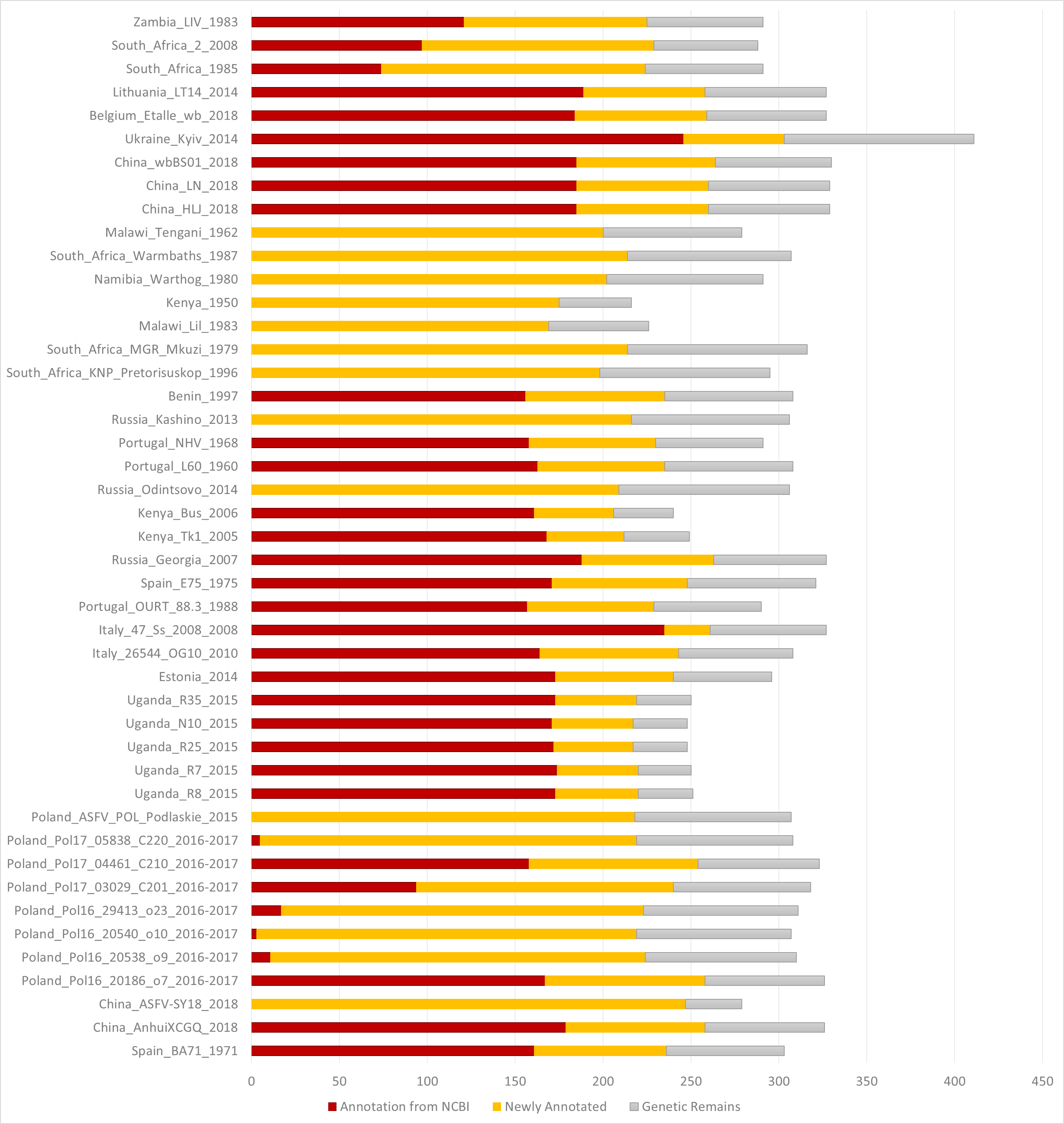
ASFVdb collected a broad range of published ASFV genome data, analyzed and identified 457 ASFV gene clusters from the genomes of all strains. Among the 457 gene clusters, 164 are conserved ORF in all strains. Reannotation of all strains’ genomes predicted 5352 genes and 3025 genetic remains. In each strain, there are 169 ~ 303 genes in average. ASFVdb did subcellular localization of the ASFV genes to display their roles in the infection process. More than half (62%) are membrane proteins. 9% are capsid proteins, providing potential targets for vaccine design. In gene ontology, the highest rate of ASFV genes enriched in membrane and the genes have catalytic activity. In population genetics test analysis, 383 genes are of significantly low Tajima’s D and significantly high composite likelihood ratio (CLR) (Rank Test, P-value<0.05), indicating these genes are possibly under positive selection recently. 811 genes are of significantly high tajima’s D value (Rank Test, P-value<0.05), and they may be in balancing selection.
Main Functions
The main search input of ASFVdb, at the center of the home page, allows users to search a gene by a general gene name, a gene accession or a description of gene’s function. In the right column of the home page, there are the list of other gene search methods in ASFVdb. Users could BLAT against one ASFV genome like the workflow in UCSC genome browser, or BLAST against the CDS sequences or protein sequences. Users could search for genes in a specific subcellular location. In the result view, ASFVdb listed the topology information and function annotation of each gene to convenient users to localize one gene in the host-virus interaction. Users also can find genes of some specific function according to the gene’s GO annotation. “Gene Clusters” lists the persistence of ASFV genes in all strains. Users can view one specific gene by clicking an item in the gene list table. Moreover, to track a list of genes more conveniently, ASFVdb offers to get gene links by inputting a list of genes’ genomic positions or a list of genes’ accession names. These operations facilitate users to do analysis in ASFVdb with a personal gene list.
Through clicking the taxon name in the tree or selecting an item in the “Gbrwoser” list, users could go to the genome browser page, where gene segments are arranged subsequently along the genome according to genes’ genomic positions. Genes annotated from the NCBI GFF file are colored in blood red as “Annotation from NCBI”. Genes annotated by mapping are colored in dark red as “Newly annotated”. “Genetic Remains” are colored in light gray. Gene Name is above each gene segment and “>” or “<” indicated a gene’s transcriptional direction. Three tracks of population genetics analysis, Pi, Theta and Tajima’s D, are listed following the gene track. To help to trace selective signatures, ASFVdb drew a top 5% line for Pi and theta, and two 5% lines in ascending and descending order for Tajima’s D. With the tool bar at the top, users can move or zoom the genome in the browser, set the focus bar of an appointed region, export the drawing of one track with gene segments and export data of one track.
Clicking one gene segment will direct to the gene’s information page. A full list of annotations include basic information (strain, gene name, description, location in the genome, Genbank Accession and full name), sequence (CDS and protein), summary (function, Uniprot accession, related Pubmed ID, related EMBL ID, corresponding Proteomes ID, related Pfam ID and correlated Interpro ID), Ontologies (GO and KEGG), subcellular location, topology (transmembrane region prediction), genomic alignment in the CDS region, multiple alignment of orthologues, gene tree of corresponding NCBI annotated or newly annotated proteins, and orthologous in strains. Texts which can link to internal or external sites are hyperlinked to facilitate view and analysis.
Methods
Data collection and processing
We downloaded the genomes of sequenced ASFV strains from NCBI. We made a unified gene list dataset and used the dataset to re-annotate ASFV genomes. Specifically, we did pairwise alignments for all annotated protein sequences. The sequences were clustered together with the identity > 90% and coverage > 70% according to previous homologues gene identification methods. We mapped these unified genes to all genomes. If a unique protein had been mapped with the highest score of identity > 50% and coverage > 80%, we considered the unique protein exists in the strain and the mapped region in the strain genome is marked. We translated the mapped region by NCBI ORFfinder and aligned all possible translations of the unique protein. We took the alignment with the highest score and checked whether its identity and coverage are both higher than 0.5. If yes, we consider the region can express proteins, if no we marked the region as ”Genetic Remains”. The genes those had been annotated in NCBI genome database marked as “Annotation from NCBI”, and the unannotated gene region with high expression possibility marked as “Newly Annotated”. At last, all genes sequenced with CD-HIT requiring identify > 90% and coverage > 70% had been clustered in 457 gene clusters.
We Blast the protein sequence of ASFV gene against the Uniprot protein database and took hit with the highest score as the best match. If the best match of one ASFV gene is of E-value <0.05, we extracted the accession of the mapped sequence as the Uniprot ID of the matched ASFV gene. With the Uniprot ID, we got external annotations of the ASFV gene, like corresponding Pubmed ID, EMBL ID, Proteomes ID, Pfam, Interpro, Gene Ontologies, KEGG and function annotations. We also Blast all ASFV protein sequences against the PDB database in the same way, and recorded the alignment results.
Subcellular localization and topology prediction
We predicted the subcellular location of ASFV proteins through MSLVP using the parameters of “one-versus-one”, “Second-tier Algorithm” and requiring similarity >90%. Gene clusters were grouped into 9 subcellular locations. We predicted transmembrane helix within proteins’ sequences using TMHMM 2.0.
Comparative genomics and population genetics analysis
We made whole genome alignment of all ASFV genomes by Mugsy, and built the phylogenetic tree by FastTree 2.1, with the parameter ‘-boot 5000’ to test the likelihood of the generated tree. W used LASTZ to do genome-genome alignments between any two ASFV strains and output the results in AXT format. For one ASFV gene, we retrieved the corresponding sequence of other strains from the genome-genome alignment results, realigned these sequences by MUSCLE, and got the genomic alignment in CDS region. According to the persistence of each gene in different strains, we also retrieved all protein counterparts of one unique gene and made the multiple alignment by MUSCLE. We also drew trees from the multiple alignment like we did for genome.
We slide along the ASFV genome with a window size of 200 bp and a step size of 50 bp. From the genome-genome alignments referred above, we retrieved sequences within the sliding window, with which we calculated Pi, Theta and tajima’s D using variscan 2.0. We wrote Perl scripts to call Allele Frequencies and used SweapFineder2 to calculate the composite likelihood ratio (CLR) with step size 50. We took the median of population genetics test statistics in the region of each gene as its corresponding value.
Web Interfaces
The web interface of ASFVdb was developed in the structure of Mysql + PHP + CodeIgniter (www.codeigniter.com) + JQuery (jquery.com), and is basing on SWAV (swav.popgenetics.net), which is an open source web application for population genetics visualization and analysis. We used MSAViewer to show the multiple alignments of orthologous CDS or proteins, and phylotree to display of the phylogenetic trees of genomes or proteins. We made changes of the source codes of the referred web application to fit the development of ASFVdb, like add links with the diagram. The search interfaces of sequence alignments are constructed basing on PHP parsing of the results from BLAT and NCBI Blast. The workflow of other search blocks, like search by molecular structure or gene names, are mostly SQL query pipelines.
